Understanding the Role of the Ubiquitin System During Cell Division
The goal of our research program is to understand how the ubiquitin-proteasome system controls cell division. To achieve this goal, we develop new chemical inhibitors of the system, and analyze effects using cell biological, biochemical and proteomic approaches.
APC/C-Dependent Protein Ubiquitination Controls Chromosome Segregation and Mitotic Exit
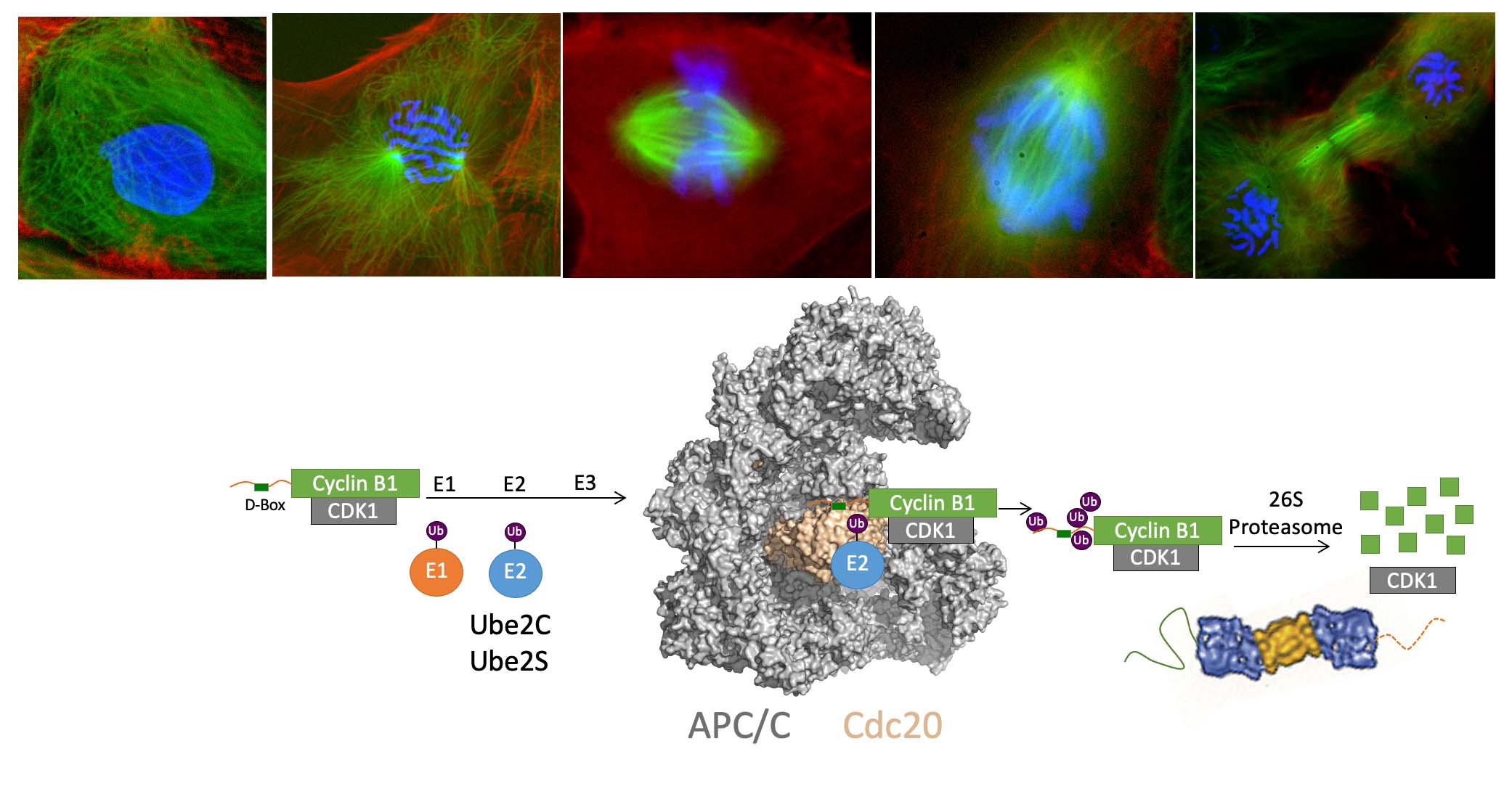
Chromosome segregation and exit from mitosis are triggered by the activation of the Anaphase-Promoting Complex/Cyclosome (APC/C), a multi-subunit ubiquitin ligase (see diagram below). The activation of the APC/C is controlled by complex mechanisms, including regulation by the spindle checkpoint, a pathway that prevents APC/C activation until all chromosomes have become properly attached to the mitotic spindle. APC/C activation requires binding of the activator protein Cdc20. The activator recruits substrates to the APC/C by binding a destruction box motif (D-box). Binding of the activator also helps the APC/C recruit an E2 enzyme (Ube2C or Ube2S) to the complex, stimulating ubiquitin transfer from the E2 to the substrate. Multiple rounds of ubiquitin transfer generate a substrate bearing multiple ubiquitin molecules, creating a signal for recognition by the proteasome, which degrades the ubiquitinated substrate. The ubiquitination of securin triggers chromosome segregation, whereas the ubiquitination of cyclin B1 (shown below) triggers CDK1 inactivation and exit from mitosis. The APC/C indeed has many substrates in mitosis and also in the G1 phase of the cell cycle, when it is activated by Cdh1 rather than Cdc20.
Identifying New Chemical Inhibitors of Cell Division
Our lab has discovered small molecule inhibitors of the APC/C that block cell division, arresting cells in metaphase. Using high-throughput screens performed in Xenopus extracts (Verma et al., 2004), our laboratory discovered two compounds that inhibit APC/C function by different mechanisms. One inhibitor, called TAME, inhibits APC/C activation by binding to the APC/C and preventing Cdc20 binding (Zeng et al., 2010; Zeng and King, 2012). The second inhibitor, called apcin, binds to Cdc20 and prevents substrate binding (Sackton et al, 2014). Using apcin together with proTAME, a cell-permeable TAME analog, synergizes to arrest cells in mitosis by disabling APC/C function (Sackton et al, 2014). When used by itself, apcin has effects that depend on the extent to which the APC/C is inhibited by the spindle checkpoint. In unperturbed cells, apcin lengthens mitotic duration by inhibiting APC/C function. However, in cells in which the spindle checkpoint is strongly activated, apcin shortens mitotic duration by interfering with the ability of the checkpoint to inhibit the APC/C. Interestingly, these opposing effects both arise from apcin binding to Cdc20 (Richeson et al, 2020). Our work provides an interesting illustration of a how a small molecule can produce different effects depending on regulatory context.
These APC/C inhibitors are now being used by many labs to study mechanisms of mitotic regulation, and they may have applications for treatment of certain cancer types (de Lange et al., 2015; Lub et al., 2016). Recently we have used APC/C inhibitors to discover new substrates of the APC/C. We found that IRS2, a component of the insulin/IGF signaling pathway, is an APC/C substrate, providing a new mechanistic link between these pathways (Manohar et al., 2020).
Mechanistic and Proteomic Studies of Ubiquitination and Deubiquitination
We are also interested in the question of how ubiquitinated proteins are targeted for degradation by the proteasome. In collaboration with the Gygi and Finley labs at HMS, we have reconstituted proteasome-dependent degradation of ubiquitinated cyclin B1 from purified components. We have used quantitative mass spectrometry to determine how the APC/C ubiquitinates its substrates (Kirkpatrick et al., 2006). We showed that cyclin B1 is ubiquitinated on multiple lysine residues, and that the APC/C and its corresponding E2 Ube2C generates short ubiquitin chains linked through lysine 48, 11 and 63. Surprisingly, we found that even a multiply monoubiquitinated form of cyclin B1 can be degraded, and that ubiquitin chains become necessary only when the number of ubiquitination sites in cyclin B1 is reduced (Dimova et al., 2012).
In collaboration with the Finley lab, we showed that the proteasome-associated deubiquitinating enzyme Ubp6 can antagonize degradation (Hanna et al., 2006) and found that the human ortholog, Usp14, has the same property (Lee et al., 2010). We have shown that Usp14 is specific for substrates that bear multiple ubiquitin chains, and that Usp14 acts until one ubiquitin chain remains (Lee et al., 2016). We also developed one of the first highly selective chemical inhibitors of a deubiquitinating enzyme, and showed that this Usp14 inhibitor can enhance the cell's ability to eliminate potentially neurotoxic or damaged proteins (Lee et al., 2010; Boselli et al., 2017). These findings may provide a new method for treatment of diseases associated with protein misfolding, such as neurodegenerative diseases.
More recently we have become interested in understanding ubiquitin dynamics and the role of deubiquitylating enzymes in Xenopus extracts. We have developed new methods to identify substrates of deubiquitylating enzymes that overcome problems associated with redundant enzyme function. We have characterized how DUBs maintain protein stability in the Xenopus system (Rossio et al., 2021). More recently, we have developed a new proteomic method to systematically identify non-proteolytic substrates of DUBs, which also enables us to directly compare the specificity and activity of these enzymes (Rossio et al, 2023).
Developing New Technologies
The lab has a longstanding interest in developing and applying new technologies to study cell division and the ubiquitin-proteasome system. We have worked closely with the Gygi lab in the Cell Biology Department to apply quantitative proteomics to study the ubiquitin system (Kirkpatrick et al., 2006; Manohar et al., 2020; Rossio et al., 2021). We have developed new technologies for high-throughput chemical screens (Walling et al., 2001; Verma et al., 2004; Lee et al., 2010) and methods for bioinformatic analysis of RNAi screens (Sigoillot et al., 2012). We apply long-term time-lapse microscopy to study cell division (Shi and King 2005; Bentley et al., 2007; Pfaff et al, 2013) and have developed new methods to automatically analyze cell division (Sigoillot et al., 2011).